Our research focuses on drug discovery and pharmaceutical sciences, with a strong emphasis on generative artificial intelligence, deep learning, chemoinformatics, and bioinformatics. We develop and apply deep learning–based and in silico modeling approaches to establish a model-driven pharmaceutical research pipeline, encompassing AI-guided target discovery from multi-omics data, molecular design and virtual screening, in silico prediction of physicochemical and ADMET properties, and model-informed clinical and translational applications. By integrating real-world clinical and manufacturing data as feedback, our work aims to accelerate drug research and development and enable data-driven decision-making in health computing.

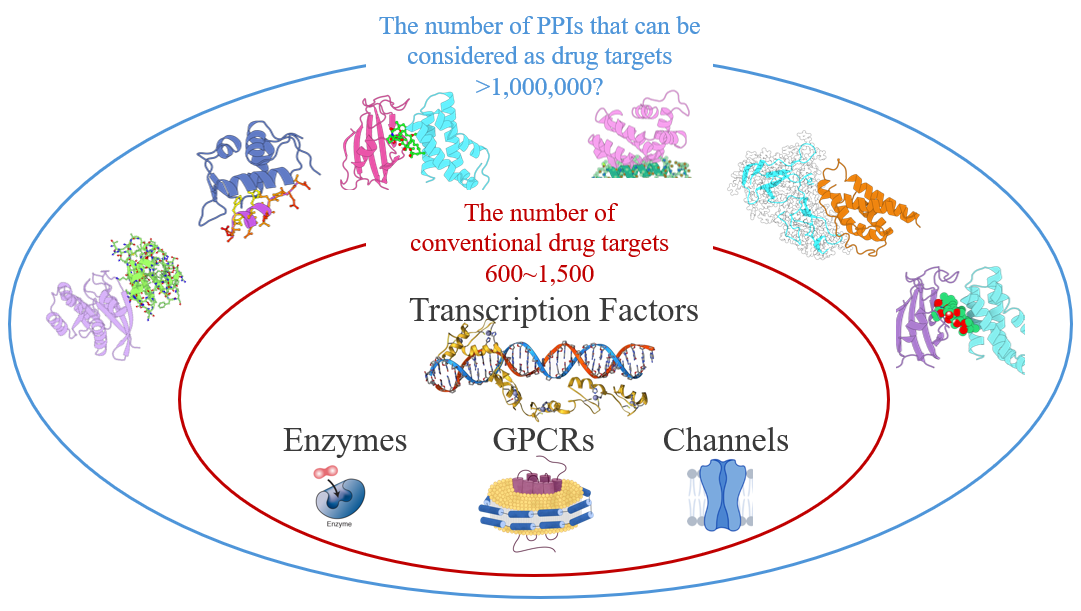
Proteins can function independently or execute specific biological roles through protein–protein interactions (PPIs). This research direction focuses on leveraging in silico computational approaches and artificial intelligence to advance PPI interaction prediction, as well as the design and screening of PPI modulators. In light of the intrinsic challenges associated with PPI interfaces—including structural complexity, limited druggability, and the frequent absence of known lead compounds—this work aims to establish a systematic AI-driven paradigm for PPI-focused interaction prediction, screening, and molecular design, thereby providing scalable computational support for the discovery of PPI-targeted therapeutics.
Biology is an inherently complex dynamical system, and capturing this complexity requires dynamic modeling approaches that can better describe and predict the emergent properties of biological systems. The conformational dynamics of proteins and protein–protein complexes lie at the heart of biological function; however, their in-depth exploration using conventional molecular dynamics (MD) simulations is often hindered by high computational costs and limited sampling efficiency. This research direction aims to integrate physics-guided generative artificial intelligence with MD simulations to enable efficient exploration of the conformational landscapes of proteins and protein–protein interactions. By training deep generative models on MD-derived trajectories, these approaches learn physically meaningful representations of protein motions and directly generate realistic conformational ensembles and trajectories. Such models complement traditional MD by accelerating sampling, uncovering previously unsampled yet physically plausible conformational states, and enabling scalable analysis of protein and protein–protein complex dynamics, thereby providing a powerful computational paradigm for elucidating structure–function relationships in biomolecular systems.